Características y alternativas de tratamiento
Síndrome de Noonan
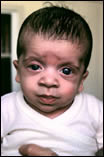
Se conoce con el nombre de síndrome de Noonan a un trastorno genético que afecta mayoritariamente a varones y que produce un anormal desarrollo en múltiples partes del cuerpo. En particular, se presentan membranas en el cuello y diferentes formas del tórax que recuerdan al síndrome de Turner (que sólo afecta a mujeres).
Estas características básicas suelen ser acompañadas de anomalías faciales, enfermedad cardíaca congénita (especialmente estenosis pulmonar), retardo mental leve en aproximadamente el 25% de los casos, pérdida auditiva variable, malformaciones genitales y bajo crecimiento.
Si bien los tratamientos son variados, se enfocan en atender las distintas afecciones por separado. Actualmente los trastornos de desarrollo se pueden tratar con hormona de crecimiento.
El síndrome de Noonan es un trastorno genético que afecta mayoritariamente a varones y produce un desarrollo anormal de múltiples partes del cuerpo. En particular, se presentan membranas en el cuello y diferentes formas del tórax que lo relacionan directamente con el síndrome de Turner, que sólo afecta a mujeres.
Diversos aspectos clínicos, cardiológicos, las enseñanzas aportadas por la ecografía fetal y finalmente los datos genéticos aclaran una parte de este síndrome, que no ha mostrado aún todos sus secretos.
Los primeros 19 casos del síndrome que reunían todas las características y síntomas hoy asignados, fueron reportados por la Dra. Jacqueline A. Noonan en el año 1968. Sin embargo en 1883 Kobilinsky describió el caso de un hombre joven que ya reunía las características básicas del síndrome y en 1928 Weissenberg contribuyó con una completa descripción de la patología.
La Dra. Noonan, en colaboración con D. A. Ehmke redescubre, años más tarde, la condición del síndrome y establece la relación del mismo con la estenosis pulmonar.
El síndrome de Noonan es un desorden heterogéneo y por ello debe ser manejado en forma multidisciplinaria. Debido al gran riesgo de complicaciones se debe hacer un diagnóstico precoz de la enfermedad y asimismo un manejo oportuno de la sintomatología.
Incidencia, causas y factores de riesgo
El síndrome de Noonan se puede heredar de una forma autosómica dominante afectando al menos a 1 de cada 2.500 niños. El gen PTPN1 es el primero que produce el síndrome de Noonan y fue descubierto recientemente, en el año 2001.
El síndrome tiene la particularidad de poseer un gran polimorfismo expresivo , en el que el signo guía más importante para el diagnóstico son los peculiares rasgos faciales a los que se asocian en combinación variable al menos uno de los siguientes: talla baja, piel redundante en cuello, implantación baja del pelo en la región occipital, deformidad esternal, cardiopatía (fundamentalmente estenosis de la válvula pulmonar), criptorquidia, y en algunos casos, déficit mental.
Todo ello en un fenotipo que va cambiando en forma evolutiva y con una fórmula citogenética normal. Cara triangular, frente amplia, epicantus, ptosis palpebral, hendiduras palpebrales hacia abajo, punta de nariz gruesa, orejas dismórficas, cuello corto.
Sin embargo, autores anteriores a la Dra. Noonan habían publicado sobre pacientes con características de tal cuadro, planteándolo como diagnóstico diferencial del síndrome de Turner, pero distinguiendo la normalidad cromosómica en ambos sexos. La virtud de Jacqueline Noonan fue que además de indicar los signos clínicos mayores, observó que la cardiopatía más frecuente era la estenosis pulmonar (17 en 19 pacientes) (Noonan 1968) frente a la cardiopatía que se presenta en el estado 45,X0 que es la coartación de la aorta.
Las anomalías que se ven con más frecuencia incluyen formación de membranas en el cuello, cambios en el esternón (generalmente un tórax hundido llamado tórax excavado), anomalías faciales y enfermedad cardíaca congénita (especialmente estenosis pulmonar ). Debido a que estas anomalías se asemejan a las del síndrome de Turner. Generalmente se presenta un retardo en la pubertad y los hombres pueden tener testículos no descendidos y un pene pequeño. Por lo general, existe una disminución de la estatura del adulto.
Síntomas
- Cuello con pliegues y de apariencia corta.
- Anomalías del esternón (tórax excavado y ocasionalmente tórax en quilla).
- Párpados caídos.
- Ojos de base amplia o inclinados hacia abajo.
- Orejas de implantación baja o de forma anormal.
- Testículos no descendidos/esterilidad.
- Retardo en la pubertad.
- Retardo mental en sólo una cuarta parte de los pacientes.
- Estatura baja.
Profundización de signos y exámenes:
Ya desde el nacimiento llaman la atención las características de la cara y cuello, pues los ojos están separados (hipertelorismo ocular), siendo los párpados superiores algo caídos o “cargados” (le-ve ptosis) y frecuentemente de forma asimétrica con hendiduras horizontales o ligeramente hacia abajo y afuera.
La raíz nasal es ancha y la punta redondeada, las orejas dismórficas por toscas, bajas, y con lóbulos prominentes y rotados hacia adelante con frecuencia. La región malar es hipoplásica. La piel de la nuca es sobrante e incluso puede apreciarse “pterigium colli”, y el pelo se extiende hacia la espalda en su implantación, sobre todo lateralmente, todo ello en un cuello que frecuentemente es corto y que aún lo parece más por el exceso de piel lateral, apreciable sobre todo al mirarlo por detrás.
Evolutivamente la cara se hace frecuentemente triangular por ser la frente amplia, la mandíbula fina, el pelo se ensortija, las cejas se arquean, los ojos se vuelven algo prominentes y las hendiduras palpebrales se hacen llamativamente “antimongoloides”. Y todo ello acaba ofreciendo una imagen de cierta tosquedad.
La deformidad esternal más común es el excavamiento inferior en un tórax habitualmente ancho, a veces con los hombros redondeados y las mamilas separadas y bajas. Se refiere en la literatura la prominencia superior “en pichón”, que es excepcional frente a la alta frecuencia de la depresión inferior.
En cuanto a los trastornos genitales, la criptorquidia se da en un 60%-75% de pacientes masculinos lo que puede llevar en formas bilaterales a favorecer la esterilidad masculina en alrededor de un 50%, con acaso menor grado de desarrollo de caracteres sexuales secundarios en la pubertad, y elevación de los niveles de gonadotropinas con bajos valores de testosterona. El pene no suele mostrar alteraciones y puede estar aumentado de tamaño. Según se halló en los estudios biópsicos testiculares, suele aparecer una disminución del índice tubular medio y de células de Sertoli llegando a la conclusión de que, además de las lesiones propias del testículo criptorquídico, hay signos de disgenesia primaria. En las mujeres, la pubertad y la fertilidad suelen ser normales. Por estas razones parece explicarse que sea habitualmente la vía materna la transmisora.
Un 11% pueden presentar anomalías tales como uréter doble, agenesia renal unilateral, por lo que la ecografía reno-ureteral está indicada.
En cuanto al desarrollo óseo, además de lo visto en el tórax, el cúbitus valgus, se aprecia en el 50%, clinobraquidactilia de algunos dedos y puntas romas con uñas pequeñas y a veces cuadradas en un 30%, con forma en cuchara y crecimiento hacia arriba frecuentemente, anomalías vertebrales en 25%.
Los exámenes pueden evidenciar en los brazos un ángulo inusual (cúbito valgo).
También puede presentarse una tendencia al sangrado, demostrado en el bajo recuento de plaquetas o en los exámenes de coagulación y en la medición de los niveles de factores específicos de coagulación en la sangre (factores XI-XIII).
Si existen signos de enfermedad cardíaca, suele recomendarse un ECG, radiografía del tórax o ecocardiograma. Se ordenan también exámenes de audición si existe algún signo de disminución de ésta. La evaluación genética puede identificar mutaciones causales en el gen PTPN11, entonces se utiliza un cariotipo para confirmar que una niña no porta en realidad el síndrome de Turner.
También se han descrito problemas de piel, manchas, edemas de causa linfática por hipoplasia, tiroiditis autoinmune, maloclusión dental y defectos oculares.
En cuanto a los valores de inteligencia, en 1/4 a 1/3 se aprecia déficit mental moderado, siendo el CI medio de 102 con valores extremos de 64 a 127. Goodman y Gorlin refieren que más de la mitad muestran retraso de ligero a CI menor de 50 y Limal y Cols indican que afecta solamente al 10%. Se ha descrito un fenotipo conductual: torpeza, tozudez, irritabilidad, con difícil comunicación y siendo caprichosos con la comida (Wood). Sharland vio que el tiempo medio de la sedestación era a los 10 meses y el de la deambulación a los 21.
Tratamiento
No se puede hablar de un tratamiento único para el síndrome de Noonan debido a la diversidad de síntomas. El tratamiento se centra en los distintos trastornos que se presentan.
Como primera medida a seguir, tras el diagnóstico neonatal fenotípico externo se impone la evaluación cardiológica. Se refiere que hasta un 6% fallecen cada año por arritmia (G.A.E.P.). Luego, la valoración cardiológica, y la observancia evolutiva del crecimiento estatoponderal y del desarrollo sicomotor. Por lo demás, como cualquier otro niño aunque pueden darse dificultades para la alimentación como la anorexia, reflujo gastro-esofágico, etc. No hay que olvidarse de la mayor frecuencia de defectos de refracción, tiroiditis, posible hipertermia maligna en caso de anestesia, hipoacusia o maloclusión dental.
El uso de la hormona de crecimiento es eficaz a corto plazo aumentando la velocidad de crecimiento, aunque sin que al parecer se incremente de forma ostensible la talla final: media de + 3.1cm (-1.1 a 6.5cm) según Kirk. No se ha constatado riesgo de cardiomiopatía hipertrófica bajo el tratamiento en el curso de 3 años de terapéutica (Cotterill).
En la pubertad, máxime los varones que tuvieron criptorquidia bilateral, han de ser evaluados gonadalmente tanto clínica (desarrollo de caracteres sexuales secundarios) como hormonalmente (determinación de valores de testosterona y gonadotropinas), dada la posibilidad de hipofunción. El tratamiento con administración de testosterona en parche diario o de depósito (cada 21 días) ha de ser individualizado para, por un lado, evitar la osteoporosis, y por otro lado que no se dé una libido potencialmente peligrosa si no se controla con una inteligencia y educación suficientes.
El pronóstico esperado depende de la extensión y gravedad de los síntomas existentes. Los pacientes pueden llevar una vida normal y, si se presenta retardo mental, generalmente es leve, dando muchas esperanzas a las familias de lograr un abordaje temprano.
Se recomienda también la participación de padres en grupos de apoyo con el fin de compartir experiencias, alternativas de abordaje y contención afectiva y profesional. En nuestro país pueden contactarse con la Asociación Civil Creciendo (http://www.creciendo.org.ar) o consultar en la asociación central de Noonan Syndrome (www.noonansyndrome.org).
Prevención
Es fundamental que aquellas personas con antecedentes familiares de síndrome de Noonan pueden acercarse a un equipo médico especializado antes de decidir tener hijos. La prevención de complicaciones, tales como enfermedad cardíaca, depende de la detección a tiempo y el cuidado continuo de un cardiólogo. Ya que no debemos olvidar que se observan defectos cardiacos congénitos en dos tercios de los pacientes: anomalías de la válvula pulmonar (50%), defectos septales auriculares (10%), defecto septal ventricular (5%) y conducto arterial persistente (3%), se han descrito otras anomalía menos comunes.
Fuentes:
- Asociación Civil Creciendo.
- Wellpath.
Otras notas
La hidrocefalia consiste en el aumento del volumen del líquido cefalorraquídeo en el interior de la cabeza del bebé en gestación, causado por un bloqueo de circulación o absorción. Distintas y variadas son las causas que pueden provocarlo, desde una...

Lo hizo el Observatorio de la Discriminación en Radio y TV, coordinado por la Afsca, que reconoció la labor del canal y de su noticiero Visión 7 en particular, por el abordaje y tratamiento de esa temática.

El síndrome alcohólico fetal es una perturbación del desarrollo que afecta a los bebés de madres que consumieron alcohol durante el embarazo, aún en las primeras semanas de gestación...